Pis'ma v ZhETF, vol.88, iss. 11, pp.762-766
© 2008 December 10
Density-functional calculation of the Coulomb repulsion and correlation strength in superconducting LaFeAsO
V. I. Anisimov, Dm. M. Korotin, S. V.Streltsov, A. V. Kozhevnikov+, J.Kunes*, A. O.Shoiikov, M. A. Koiotin1^ Institute of Metal Physics RAS, 620041 Yekaterinburg GSP-170, Russia +Joint Institute for Computational Sciences, Oak Ridge National Laboratory Oak, TN 37831-6173 Ridge, USA
* Theoretical Physics III, Center for Electronic Correlations and Magnetism, Institute of Physics, University of Augsburg, 86135
Augsburg, Germany
Submitted 8 October 2008 Resubmitted 5 November 2008
Constrained density functional theory scheme in Wannier functions formalism has been used to calculate Coulomb repulsion V and Hund's exchange J parameters for Fe-3d electrons in LaFeAsO. Results strongly depend on the basis set. When 0-2p, As-4p, and Fe-3d orbitals are included, computation results in U=3^4 eV. With the basis set restricted to Fe-3d orbitals only, computation gives parameters corresponding to F°=0.8 eV, J=0.5 eV. Local Density Approximation combined with Dynamical Mean-Field Theory calculation with these parameters results in weakly correlated electronic structure.
PACS: 71.45.Gm, 74.25.Jb
Following the discovery of high-Tc superconductivity in iron oxypnictide LaOi-^.Fj.FeAs [1], a question of the influence of electronic correlation effects on the normal and superconducting properties of LaFeAsO has arisen. In striking similarity with high-Tc cuprates, un-doped material LaFeAsO is not superconducting with antiferromagnetic commensurate spin density wave developing below 150 K [2]. Only when electrons (or holes) are added to the system via doping, antiferromagnetism is suppressed and superconductivity appears. As it is generally accepted that Coulomb correlations between copper 3d electrons are responsible for cuprates anomalous properties, it is tempting to suggest that the same is true for iron 3d electrons in LaFeAsO.
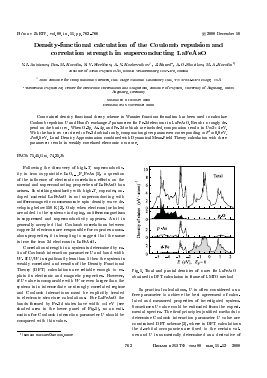
Correlation strength in a system is determined by ratio of Coulomb interaction parameter U and band width W. If U/W is significantly less than 1 then the system is weakly correlated and results of the Density Functional Theory (DFT) calculations are reliable enough to explain its electronic and magnetic properties. However, if U value is comparable with W or even larger than the system is in intermediate or strongly correlated regime and Coulomb interactions must be explicitly treated in electronic structure calculations. For LaFeAsO the bands formed by Fe-3<i states have width «4 eV (see shaded area in the lower panel of Fig.l), so an estimation for Coulomb interaction parameter U should be compared with this value.
e-mail: michael@korotin.name
a
o
■a
I
O
S
Q
-4 -3 -2 -1 0 E (eV), Ef= 0
Fig.l. Total and partial densities of states for LaFeAsO obtained in DFT calculation in frame of LMTO method
In practical calculations, U is often considered as a free parameter to achieve the best agreement of calculated and measured properties of investigated system. Sometimes U value could be estimated from the experimental spectra. The first principles justified methods to determine Coulomb interaction parameter U value are constrained DFT scheme [3], where in DFT calculations the d-orbital occupancies are fixed to the certain values and U is numerically determined as a derivative of
762
IlHCbMa b ?K3T<D tom 88 btra.11-12 2008
d-orbital energy over its occupancy and Random Phase Approximation (RPA) method, where screened Coulomb interaction between d-electrons is calculated via perturbation theory [4]. In Ref. [5] it was proposed to use in LaFeAsO U 4 eV obtained in RPA calculations for metallic iron [6].
This value for Coulomb parameter (with Hund's exchange parameter J=0.7 eV) was used in Dynamical Mean-Field Theory (DMFT) [7] calculations for LaFeAsO [5, 8, 9]. Results of these works show iron 3d electrons being in intermediate or strongly correlated regime, as it is natural to be expected for Coulomb parameter value V 4 eV and Fe-3d band width «4 eV.
The most direct way to estimate correlation effects strength in a system under consideration is to compare the experimental spectra with densities of states (DOS) obtained in DFT calculations. For strongly correlated materials additional features in the experimental photoemission and absorption spectra appear that are interpreted as lower and upper Hubbard bands absent in the DFT DOS. If no such additional features are observed and DOS obtained in DFT calculations satisfactorily describe the experimental spectra then the material is in weakly correlated regime.
LaFeAsO was studied by soft X-ray absorption and emission spectroscopy [10], X-ray absorption spectroscopy (O iT-edge) [11], and photoemission spectroscopy [12]. In all these works the conclusion was that DOS obtained in DFT calculations gave good agreement with the experimental spectra and the estimations for Coulomb parameter value are U <1 eV [11]. That contradiction with results of the DMFT calculations [5, 9, 8] shows that first principles calculation of Coulomb interaction parameter U value for LaFeAsO is needed to determine the correlation effects strength in this material. Results of such calculations by constrained DFT calculations are reported in the present work. We have obtained the value U <1 eV for Fe-3d band that agrees with the estimates from spectroscopy. Recently the RPA calculations for Coulomb interaction parameter U in LaFeAsO were reported, where U was estimated as 1.8^2.7 eV [13].
It is important to note that Coulomb interaction parameter U value depends on the model where it will be used and, more precisely, on the choice of the orbital set that is taken explicitly into account in the model. For example, in constrained DFT calculations for high-Tc cuprates the resulting U value for Cu d-shell was found between 8 and 10 eV [14]. The U value in this range was used in cluster calculations where all Cu d-orbitals and p-orbitals of neighboring oxygens were taken into account and calculated spectra agree well with experimen-
tal data [15]. However, in one band model, where only x2 — y2-orbital per cooper atom is explicitly included in the calculations, the U value giving good agreement with experimental data falls down to 2.5 : 3.6 eV [16], that is 3-4 times smaller than constrained DFT value.
The same situation occurs for titanium and vanadium oxides: the U value from constrained DFT calculations is «6 eV and cluster calculations where all d-orbitals and p-orbitals of neighboring oxygens were taken into account with U close to this value gave good agreement between calculated and experimental spectra [17]. However, in the model where only partially filled i2S-orbitals are included, much smaller U value (corresponding to Slater integral i'°=3.5 eV) gives the results in agreement with experimental data [18].
It is interesting that such a small U value can be obtained in constrained DFT calculations for titanates and vanadates where only i2s-orbital occupancies are fixed while all the other states (es-orbitals of vanadium and p-orbitals of oxygens) are allowed to relax in self-consistent iterations [19, 18]. So the calculation scheme used in constrained DFT (the set of the orbitals with fixed occupancies) should be consistent with basis set of the model where the calculated U value will be used.
Another source of uncertainty in constrained DFT calculation scheme is a definition of atomic orbitals whose occupancies are fixed and energy calculated. In some DFT methods, like Linearized Muffin-Tin Orbitals (LMTO) [20], these orbitals could be identified with LMTO. However, in other DFT calculation schemes, where plane waves are used as a basis, like in pseudopotential method [21] one should use more general definition for localized atomic like orbitals such as Wannier functions (WFs) [22]. The practical way to calculate WFs for specific materials using projection of atomic orbitals on Bloch functions was developed in [23].
In Fig.l the total and partial DOS for LaFeAsO obtained in LMTO calculations with crystal structure data taken from [1] and default set of parameters as realized in Stuttgart TB-LMTO-ASA program are shown. The results are very similar to previous ones (e.g., [24]). Crystal field splitting for Fe-3d orbitals in this material is rather weak (Ac/=0.25 eV) and all five d-orbitals of iron form common band in the energy region (—2, +2) eV relative to the Fermi level (see grey region on the bottom panel in Fig.l). There is a strong hybridization of iron i2s-orbitals with p-orbitals of arsenic atoms which form nearest neighbors tetrahedron around iron ion. This effect becomes apparent in the energy interval (—3, —2) eV (white region on the bottom panel in Fig.l) where band formed by p-orbitals of arsenic is situated. More weak hybridization with oxygen p states appears
764
V. I. Anisimov, Dm. M. Korotin, S. V. Streltsov et a1.
in the (—5.5, —3) eV energy window (black region on the bottom panel in Fig.l).
We have calculated Coulomb interaction U and Hund's exchange J parameters for WFs basis set via constrained DFT procedure with fixed occupancies for WFs of d-symmetry. For this purpose we have used two calculation schemes based on the different DFT methods. One of them involves linearized muffin-tin orbitals produced by the TB-LMTO-ASA code [20]; corresponding WFs calculation procedure is described in detail in [25]. The second one is based on the plane waves obtained within the pseudopotential plane-wave method PWSCF, as implemented in the Quantum ESPRESSO package [21], and described in detail in [26] (including choice of pseudopotentials and cut-off energy). The difference between the results of these two schemes could give an estimation for the error of U and J determination.
The WFs are defined by the choice of Bloch functions Hilbert space and by a set of trial localized o
Для дальнейшего прочтения статьи необходимо приобрести полный текст. Статьи высылаются в формате PDF на указанную при оплате почту. Время доставки составляет менее 10 минут. Стоимость одной статьи — 150 рублей.